






FDA 征求对 3D 打印医疗器材的监管提供反馈
2022-01-30
根据3D科学谷的了解,美国食品和药物管理局 (FDA) 已就其设计的面向未来的监管框架公开征求意见,以确保 3D 打印医疗器材的质量,以尽可能完善其即将出台的 3D 打印医疗器材法规。

为未来监管提供指导
为临床采用铺平道路
FDA 以讨论文件的形式发布,不仅概述了护理点 3D 打印的管理方式,而且确定了其最终用途的挑战并提供了修订后的潜在规则。FDA 表示这些建议不是作为指导,而是旨在“提出问题”,因此现在正在征求医疗 3D 打印行业的反馈,这将有助于为未来的监管提供信息。

根据 FDA 的说法,在医院和手术中采用 3D 打印可以快速生产特定于患者的器材,如解剖模型以及其他即时医疗器械。FDA强调了3D打印技术在帮助解决供应链问题方面的作用,例如在大流行的早期阶段,全球范围内出现的治疗和预防 COVID-19 方面的医疗资源的短缺。
FDA的讨论文件侧重于设备和放射健康中心 (CDRH) 对其监管拥有管辖权的设备。过去,FDA的立法已被用于从 Onkos Surgical 的 3D 打印 BioGrip 项圈到隶属于金属3D打印机制造商Desktop Metal的Desktop Health推出的Flexcera Base树脂用于3D打印牙齿修复体,为这些应用的临床采用铺平了道路。

然而,FDA 也承认大规模推广此类设备存在挑战,包括供应商不太可能拥有与传统制造商相同水平的生产知识。因此,为了确保仪器保持安全和适用于目的,FDA现在正在寻求后者关于所谓的“监管方法的初步大纲”的反馈。
PhonoGraft 3D 打印仿生植入物(如图)的创造者目前正在寻求 FDA 对其器材的批准。

正在 3D 打印的 PhonoGraft 仿生植入物。© Wyss 研究所
在其讨论文件中,FDA 强调了 3D 打印机制造商与护理设备生产商的技术和培训之间的差异如何导致最终产品质量的不同。这些不匹配的范围从内部流程和临床实践指南等方面,一直到后处理设备的可用性,在某些情况下这可能对安全至关重要。
为了解决这些潜在的缺陷,已经概述了一个五步基础,在此基础上可以建立一个修改后的方法来管理医护点的3D 打印设备。总体而言,FDA 强调打算在该过程中“采用基于风险的方法”,并考虑到一些医疗保健提供者可能无法在日常职责之外管理这些风险。
FDA还表示,器材制造地点的变化不应影响其满足规范的能力,而当临床医生采用任何先进技术时,培训应就位。有趣的是,FDA 也呼吁采用“负担最少的方法”来实现这一目标,并建议“依赖现有标准和流程”可能是这方面的关键。
为了进一步激发关于未来监管可能是什么样子的辩论,FDA 添加了三个假设,首先,医疗保健提供者在现场安装3D打印机,其次,医院或手术室也采用相关流程,最后,与附近的服务商签约生产医疗器材并交付。
在 FDA 的看来,每种情况都提出了关于不良反应报告的问题,如何在监管条款中适应设计的变化以及哪些器材可以归类为“低风险”。
最终,FDA 表示“所有利益相关者之间的沟通”是“为技术增长和发展创造环境”的关键。3D 打印发展如此之快,以至于当前的监管在未来几年可能不跟不上其发展的脚步,因此FDA旨在创建一个修订后的规则集,以“平衡创新与监管监督”。
重要的是,FDA这份讨论文件的发布旨在促进讨论并征求公众的反馈意见,这些反馈将有助于为在护理点、患者的个性化护理和该领域的新创新方面制定适当的 3D 打印监管方法奠定基础。
走上正轨的3D打印医疗器械
根据3D科学谷的了解,定制式医疗器械在我国也得到了应用发展,2020年1月1日,《定制式医疗器械监督管理规定(试行)》正式实施。该规定使得3D打印定制化骨科植入物在接受监管的过程中有依据可寻,促进3D打印定制化骨科植入物技术在临床中的应用发展。
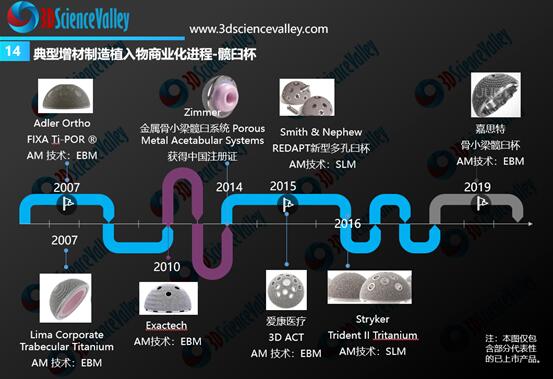
根据3D科学谷的市场研究,3D打印技术在骨科植入物制造中的应用可以分为:标准化植入物、患者匹配型植入物和定制化植入物。根据规定中的定义,其中为解决传统方式无法解决的临床问题的3D打印定制化植入物,属于《定制式医疗器械监督管理规定(试行)》的适用范围。
采用备案制进行监管
与标准化批量生产的医疗器械不同,定制式医疗器械无需经过漫长的实验与审批管理周期,而是按规定实行上市前备案管理。
定制式医疗器械生产企业与医疗机构共同作为备案人,在生产、使用定制式医疗器械前应当向医疗器械生产企业所在地(进口产品为代理人所在地)省、自治区、直辖市药品监督管理部门备案。从风险控制的角度出发,定制式医疗器械不得委托生产,备案人应当具备相应条件。
设计加工
在保护患者隐私的情况下,生产企业应当将定制式医疗器械产品设计环节延伸到医疗机构。
定制式医疗器械研制、生产除符合医疗器械生产质量管理规范及相关附录要求外,还应当满足以下特殊要求:1.人员;2.设计开发;3.质量控制;4.追溯管理。
使用数量达到上市前审批要求之后
规定指出,当定制式医疗器械临床使用病例数及前期研究能够达到上市前审批要求时,相关生产企业应当按照《医疗器械注册管理办法》《体外诊断试剂注册管理办法》规定,申报注册或者办理备案。符合伦理准则且真实、准确、完整、可溯源的临床使用数据,可以作为临床评价资料用于注册申报。
例如,金属3D打印定制式颈椎融合体,在临床应用一定例数、产品基本定型后,可以作为患者匹配医疗器械申报注册。患者匹配型植入物,属于常见疾病植入物的定制化解决方案,进入到这一阶段时,3D打印的应用将属于植入物产品批量定制化生产应用。
有关规定中公布的更多定制式医疗器械监督管理细则,请参考《定制式医疗器械监督管理规定(试行)》。3D打印技术在骨科植入物制造领域的应用,请参考3D打印与骨科植入物白皮书2.0。
来源:3D科学谷


