






法规政策
- 2025.04.01
上海医疗器械展|创新医疗器械的审批流程是怎样的?
上海医疗器械展带你了解创新医疗器械的审批流程。

阅读更多 - 2025.03.25
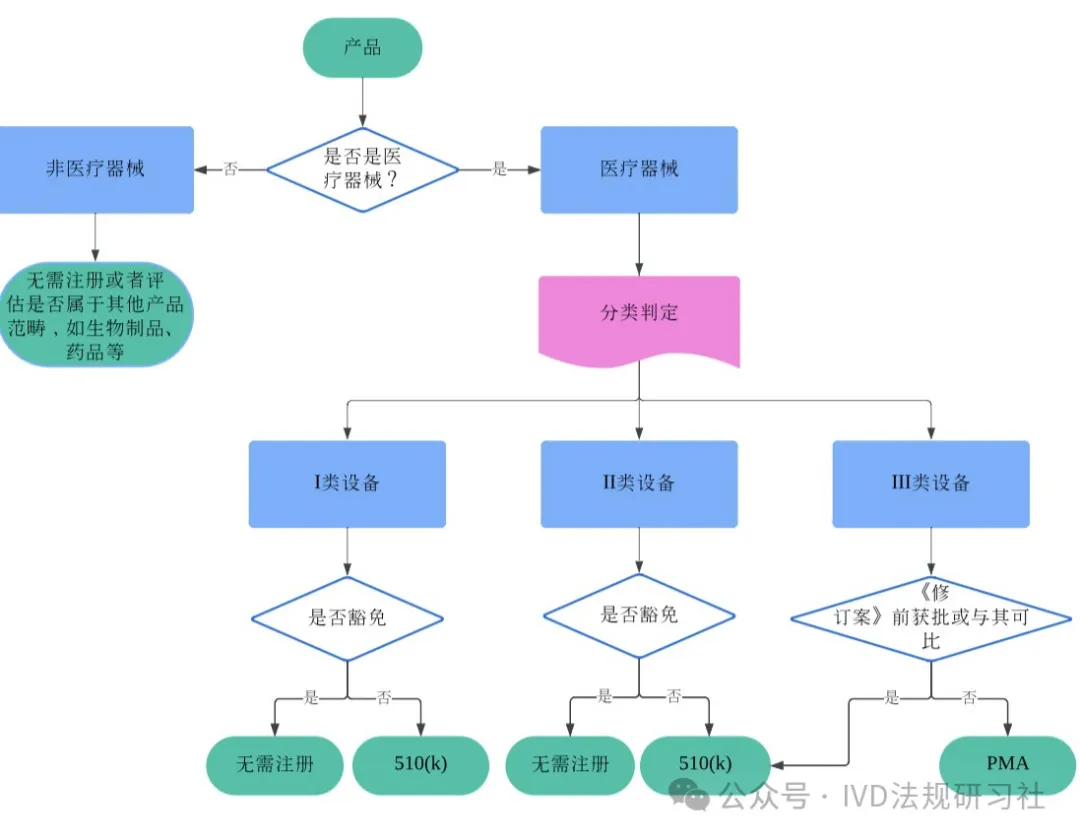
中国医疗器械博览会|FDA|一文读懂FDA医疗器械分类和注册途径选择
中国医疗器械博览会了解到,确定产品类别是启动医疗器械注册的第一步。了解FDA医疗器械的分类法规至关重要,不仅有助于制造商顺利将医疗器械产品引入美国市场,避免因分类错误导致的对注册时限、费用和资源的影响和损耗,以及可能面临的被扣货/拒绝入境/列入黑名单等风险,同时也有助于注册从业者学习和比较不同监管机构要求上的异同,拓展法规知识。
阅读更多 - 2025.02.26
上海医疗器械展会|人工智能医疗器械注册:新技术带来的新挑战
上海医疗器械展会了解到,近年来,人工智能(AI)技术在医疗器械领域的应用迅速发展。从影像诊断到手术机器人,再到智能健康管理设备,AI医疗器械正成为医疗智能化的关键驱动力。

阅读更多 -
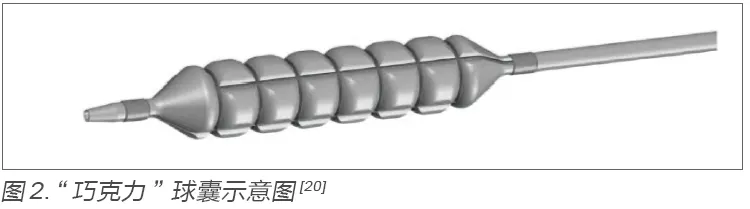
上海医疗器械展|【学术分享】血管内特殊球囊审评思考
上海医疗器械展了解到,血管内应用的球囊扩张导管一般由导管管体、球囊、不透射线标记、导管座等结构组成。普通球囊扩张血管时血管壁受力不均,导致血管内膜/ 斑块无序撕裂,用于中、重度冠状动脉钙化病变扩张成功率低而并发症发生率高,用于股浅动脉病变严重夹层(C-F型)发生率达42%、支架补救率达74.2%[1,2]。
阅读更多 - 2025.02.12
国际医疗器械展览会|FDA最新发布“人工智能设备指南草案”解读
2025年1月7日,美国食品药品监督管理局FDA发布了“针对人工智能医疗器械开发商的综合指导草案”指南——《人工智能设备软件功能:生命周期管理及上市提交建议草案》。

阅读更多 - 2025.02.08
中国医疗器械展|中国医疗器械UDI法规、要求及实例解析
在医疗器械行业的发展进程中,医疗器械唯一标识(UDI)的应用已成为提升监管效能、保障医疗安全的关键举措。

阅读更多 -
中国医疗器械展|国内医疗器械临床评价路径有哪些
医疗器械的临床评价是确保其安全、有效并得以合法上市的关键环节。中国医疗器械展了解到,在国内,主要有以下三种临床评价路径。
阅读更多 - 2025.01.02
2025医疗器械展会Medtec一文读懂美国FDA和欧盟MDR医疗器械法规异同!
医疗器械监管的世界是复杂的,欧盟和美国在证明等同性的方法上存在显著差异。对于有出海意向的国内医疗器械企业来说,美国和欧盟地区应该是首选的两个市场。而器械制造商对于目标市场地区的监管法令是一定要做足功课的。但与此同时,地区与地区之间在监管法令方面势必会存在一些差异,这也就需要制造商们“对号入座”,选择适合自己的途径,以及合理避开一些障碍。

阅读更多 - 2024.11.21
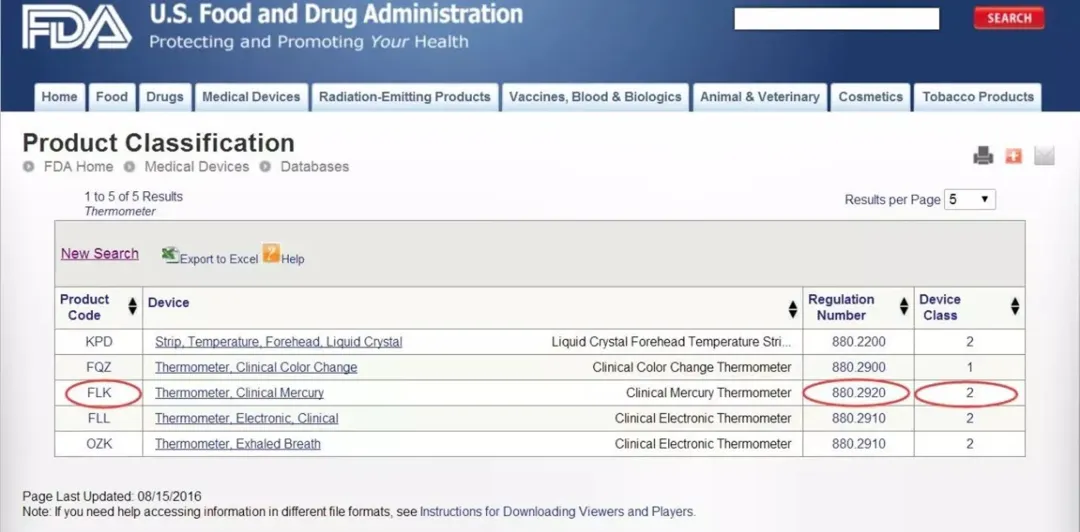
2025上海高端医疗设备展谈欧盟MDR与美国FDA医疗器械分类方法详解
阅读更多 - 2024.11.15
有源医疗设备展解读12部门发文,创新医疗器械迎利好
阅读更多












