






医疗器械生产过程中包装相关环节的质量控制
2020-10-29
医疗器械的包装从采购到最终装入产品后出厂,整个过程都应在受控的情况下进行。控制的依据为《医疗器械生产质量管理规范》,控制的标准则需根据产品的实际情况而决定,但包装的生产环境级别不能低于所装产品要求的环 境级别,且包装的传递和使用应能够保证产品的质量。
1 包装及其生产环境相关的法规要求
为加强对医疗器械的监管力度,提升医疗器械生产企业的质量管理水平,保障医疗器械产品的安全有效,原国家食品药品监督管理总局于2015年7月17日发布了无菌医疗器械、植入性医疗器械和体外诊断试剂3类产品的《医疗器械生产质量管理规范》附录,对这3类医疗器械产品作出了特殊要求。
1.1 无菌医疗器械
对于无菌医疗器械,需根据产品质量要求确定初包装材料的初始污染菌和微粒污染可接受的水平。植入和介入血管内的无菌医疗器械的初包装和封口的生产区域洁净度应不低于10000级;与血液、骨髓腔或非自然腔道直接或间接接触的无菌医疗器械的初包装和封口的生产区域洁净度应不低于100000级;与人体损伤表面和黏膜接触的无菌医疗器械的初包装和封口的生产区域洁净度应不低于300000级;与无菌医疗器械的使用表面直接接触、无需清洁处理即使用的初包装材料,其生产环境洁净度级别的设置应与产品生产环境的洁净度级别相同;若初包装材料不与无菌医疗器械使用表面直接接触,应在不低于300000级的洁净室(区)内生产。
1.2 植入性医疗器械
主要与骨接触的植入性无菌医疗器械的初包装和封口的生产区域洁净度应不低于100000级;主要与组织和组织液接触的植入性无菌医疗器械的初包装和封口的生产区域洁净度应不低于100000级;主要与血液接触的植入性无菌医疗器械的初包装和封口的生产区域洁净度应不低于10000级;与人体损伤表面和黏膜接触的植入性无菌医疗器械的初包装和封口的生产区域洁净度应不低于300000级;若初包装材料不与植入性无菌医疗器械使用表面直接接触,应在不低于300000级的洁净室(区)内生产。
1.3 体外诊断试剂
体外诊断试剂产品内包装的生产区域洁净度应不低于100000级,洁净度级别的控制标准应符合《无菌医疗器械生产管理规范》中的要求,该管理规范对洁净室(区)的设计、建造和监测都有规定,洁净室(区)的控制指标[4]见表1;另外,可以通过测试表中的指标,验证生产区域是否达到附录中要求的洁净级别。
表1 无菌医疗器具洁净室(区)的环境要求 
2 包装在医疗器械生产过程中的工艺流程
包装在医疗器械生产过程中的工艺流程可以简述为以下几步:(1)在包装生产企业生产加工;(2)通过销售采购程序进入医疗器械生产企业;(3)使用前脱去外包装进入生产洁净区;(4)在洁净区内暂存,存放地点可以是中间品库或生产线;(5)包装使用,采用包装机对产品进行包装,此步骤会用到工艺用气;(6)包装完成后传递到洁净区外,传递方式可以选择传递窗或物流缓冲;(7)在非洁净区进行外包装;(8)进行灭菌处理,灭菌后对包装进行验证。具体工艺流程见图1。
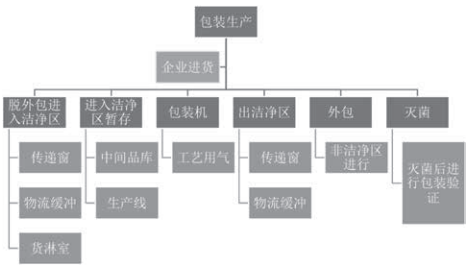
图1 包装在医疗器械生产过程中的工艺流程
3 包装在各个生产工艺流程中的控制要点
3.1 采购和进货验收
包装采购时,需考察供应商的资质和生产环境,保证环境级别与包装的要求相适应;同时,要求供应商提供包装材料的性能报告,保证包装材料质量的稳定性和可靠性;对于洁净级别要求较高的包装,应采用多层包装的形式, 以保证其在运输和储存过程中能够保持洁净等级。无菌产品的包装在采购进厂后,需检测初始污染菌和微粒污染水平,以控制包装的初始状态,减少对产品的污染。若初始污染菌过高,则会在传递和包装操作中污染洁净区和产品,因此,必须提高灭菌条件以达到无菌水平,但较大灭菌剂量可能会对产品的性能、质量或效期产生影响。微粒的污染水平与初始污染菌类似,对于植入类或输液器类接触人体的医疗器械,微粒进入人体的风险大,因此,应控制微粒的数量,尤其对于采购后直接使用,不再进行清洗的包装,更需注意微粒的控制,防止其污染到产品。
3.2 脱外包装进入洁净区
包装进入洁净区有3种方式,分别为传递窗、物流缓冲(闸间)和货淋室,针对选择何种方式进入洁净区,并无特别的规定,但可以根据包装物料的体积和生产区域的设施、设备进行选择。当包装物料较少时,选择 传递窗的方式较为简单便捷;当包装物料较多但是不会超过物流缓冲间的容积时,可以选择物流缓冲的方式,以实现一次传递;当包装物料较多且超过物流缓冲间的容积时,可以选择货淋室的方式进行传递,货淋室可以进行连续分次传递,但是需要包装物料在货淋室停留足够的时间。
3.2.1 传递窗
传递窗[5]按照使用功能分为基本型、净化型、消毒型、负压型、气密型5类。用于包装传递时推荐选用净化型,可以更好地防止传递的物品污染洁净环境。传递窗安装后, 需要确认其外观、喷口中心风速、换气次数、洁净度、压差、噪声、门互锁功能和电气安全等性能。传递窗的日常维护包括记录紫外灯的寿命、检测传递窗内的洁净度、检查窗门的密封性和互锁的有效性;另外,要正确使用传递窗,避免传递窗两侧窗门同时开启。
3.2.2 物流缓冲
当包装体积较大或数量较多时,需要从物流缓冲间进入洁净区,物流缓冲间的压差和换气次数需符合要求,避免对包装造成污染;另外,需要验证包装在物流缓冲间内放置的位置和时间,保证包装达到预期的净化效果后方可进入洁净区。
3.2.3 货淋室
货淋室[6]的性能也需要经过检测确认,检测项目包括洁净度、风速、喷口、噪声、照度等。日常使用过程中,应定期更换过滤器,日光灯和风罩发生损坏时应及时更换,以确保设备正常工作。
3.3 在洁净区内暂存
包装进入洁净区后,会有两种储存情况:(1)不会立即进行包装操作,而是放入中间品库暂存;(2)直接到内包车间进行产品的包装操作。放入中间品库储存,一般时间较长,应注意控制储存环境,尤其是温湿度,长时间处于高温高湿的环境中,会影响包装材料和包装封口的状态,只有在适宜的温湿度下储存,才能更好地保持包装的进厂状态;此外,应记录和标识包装进入中间品库的时间和存放的位置,遵循先进先出的原则进行取用,防止不同批次包装的混用错用。直接进入生产线生产的包装,分为两种情况:(1)所有包装当日全部用完;(2)包装部分用完,有剩余包装要在下一班次生产时继续使用。当日全部用完的情况,风险较低,只需注意生产过程的控制即可;有剩余包装要再次使用的情况,风险较高,此时包装已是开启待用状态,容易受到污染,两个班次之间间隔时间的长短会对包装产生不同的影响,间隔时间越短影响越小,如果间隔时间≥1周,应该进行验证后再投入使用。
3.4 使用包装机进行包装
气动包装机是医疗器械产品内包装常用的包装方式,设备运行过程中会用到工艺用气,应对工艺用气的性能进行检测,保证其不会污染包装和产品;工艺用气如果在洁净区内有排放,还应保证排放出的工艺用气不会污染洁净区内的环境,对于工艺用气的质量控制可以参考相关的指南。
3.5 流出洁净区
内包装完成后,会传递到洁净区外进行外包装,传递方式一般使用传递窗或物流缓冲,控制要点与包装流入时相同;但是,在传递出洁净区的环节,包装内装有产品,特别是形状带有尖锐部分的产品,传递过程中应注意保持包装完整,防止包装表面发生破损,从而造成污染。
3.6 外包装后灭菌
在非洁净区内,产品完成外包装后将按照预定的方式进行灭菌,灭菌完成后产品的整个生产过程结束;灭菌后的包装需要进行再确认,防止灭菌过程对包装材料或封口造成不良的影响。
4 小结
包装作为医疗器械生产过程中的重要环节,既要满足《医疗器械生产质量管理规范》的要求,也要满足对于包装本身的要求,才能保证最终产品的要求。无菌医疗器械、植入性医疗器械和体外诊断试剂是特殊的医疗器械,因此,对其包装的要求也更为严格,以便为产品的合格生产提供保证。
来源:医疗装备杂志


