






创新医疗器械如何跨越商业化陷阱?
2019-10-16
一、前言:医疗器械“因创新而失败”的怪相
医疗器械,为何创新维艰?
创新医疗器械实现产品化,只是万里长征第一步。当人们历经千难万险,为翻过“产品化”这座大山欢欣鼓舞之时,极目远眺,却又发现,商业化,将是一座更加难以逾越的险峰。
这条路难到什么程度?坊间早有戏言,当下搞医疗器械创新,“就是跟钱过不去”。
资本的嗅觉从来敏锐,而资本追捧的方向,足以管中窥豹。
诚然,在医疗器械投资领域,创新是永不过时的焦点。但最近几年的投融资实践表明,越创新的医疗器械,资本反而越谨慎。谨慎的原因,无外乎商业化难度太高,用时过长,不符合资本对收益率和回报周期的追求。相形之下,“进口替代”这一主题,一直稳居医疗器械投融资的热点。假如某种医疗器械,技术已在海外市场发展成熟,产品已取得CE或FDA注册,临床上已有一定规模的应用,则最具“进口替代”的商业潜力。相关背景的创业人士,无论是直接将类似技术拿回国内市场“重走一遍流程”,还是在进口技术基础上本土化研发“Me-better”产品,都极具潜力成为投资人众星捧月的明星项目。
而对于创新医疗器械,企业不仅要面对“创”背后的困难,还要应付“新”带来的挑战。最令人头痛的是,正因为这些挑战由“新”而来,即使企业早做准备,仍难以对挑战的发生规律、关键节点和影响程度有全面认识。这给创新医疗器械的商业化,带来极大不确定性。
二、创新医疗器械三大商业化陷阱
具体而言,创新医疗器械商业化,主要面临着以下三大“不确定性陷阱”:
-
陷阱一,产品注册冗长
-
陷阱二,物价审核困难
-
陷阱三,临床接纳迟缓
陷阱一:产品注册冗长
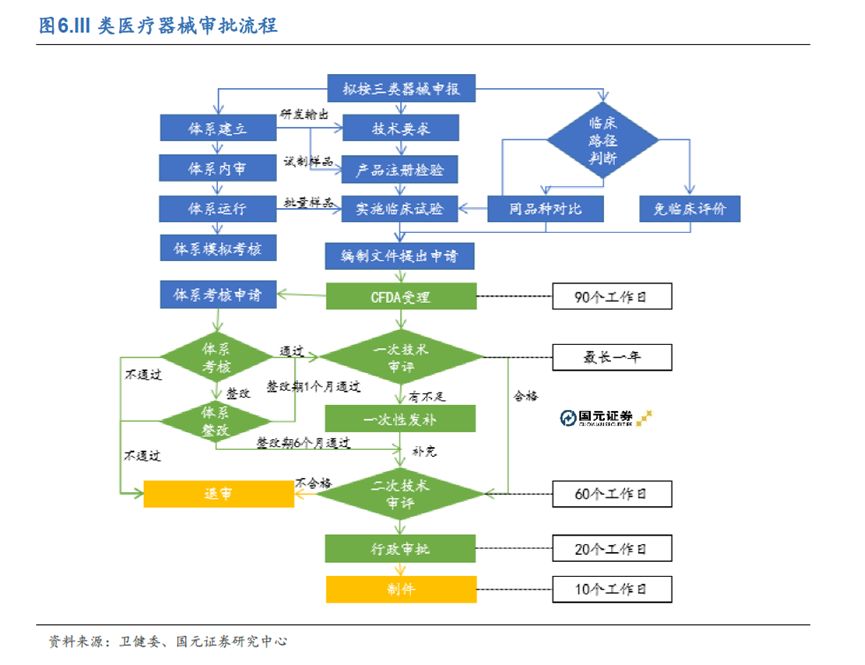
中国医疗器械注册,经过多年监管收紧后,可谓“世界最严”,并以三类器械尤甚。而这也正是大部分创新医疗器械的归口分类。3-5年的时间成本,动辄千万的资金投入,几乎已成为三类医疗器械注册的“标配”。
纵观三类器械注册全流程,可分为注册检验、临床评价、注册申报三步。过去几年,政府在积弊最多、诟病最重的“注册申报”阶段大刀阔斧,力推“创新医疗器械特别审查程序”,成功将创新器械在该阶段的平均用时,从约180工作日缩短到至约100工作日(不计发补),成效显著。但是,在剩余的“注册检验”和“临床评价”两步骤,仍有肉眼可见的挑战,阻碍着创新医疗器械商业化的顺利推进。
注册检验阶段,由于检验机构缺乏经验、动力不足,创新医疗器械很容易掉入注册检验的“堰塞湖”。
理论上,注册检验正常所需时间是约150工作日(若无整改)。然而,由于近期一系列政策变化,创新医疗器械,注册检验所需时间很可能大幅增长。一方面,作为国家取消行政事业收费的举措之一,国务院在2017年3月取消了医疗器械注册检验收费。由于全国仅有50余家有资质的医疗器械检验机构,且机构们对新政缺乏足够预案,导至企业送检大排长龙,检验时间大幅拉长;另一方面,由于创新医疗器械技术存在新颖性,检验机构很有可能会出现“人员存在知识盲区,单位缺乏专门设备”的情况。此情形下,已经对暴涨的工作量力不从心的检验机构,很难有时间和动力针对该创新器械进行“定向学习”和“定制服务”。检验进度出现进一步拖延,再正常不过。
在此阶段遇阻的典型案例,莫过于某中日合资人工心脏企业。最近,该企业的人工心脏,作为首个国产产品由NMPA获批上市的消息,在医疗器械行业朋友圈疯狂刷屏,一时风光无量。但风光背后,该企业产品的注册检验之路,却充满了心酸和无奈。
作为2014年从日本技术引进的创新医疗器械,该产品在引进中国前,已被日本监管机构审批上市,并获得了美国FDA的临床试验豁免,随即成功上市。引进中国后,该技术也在2015年底获得了CFDA创新医疗器械特别审批资格。但是,由于当时国内人工心脏领域尚属空白,缺乏检验经验和检验设备,又恰逢国内检验机构引入新的检验标准,注册进度一直卡在检验这一步,一拖竟是数年。
该公司认为,其人工心脏产品,属于已在发达国家市场获得验证的成熟技术,理应按照国际惯例,在国内注册检验阶段,减免在国际市场已经做过的测试项目。尽管公司意见获得了相关专家的认可,但检验机构的反馈,却一直悬而未决,进展缓慢。截至2018年,公司已经烧了约8000余万元在产品引进和注册阶段,但因为检验进度一拖再拖,一度传出了公司濒临破产、合作濒临中断的消息。2018年1月,在注册检验阶段卡了近两年半以后,该产品终于了通过相关检验,获得了临床批件。
临床评价阶段,由于创新器械几乎无法豁免临床试验,使得临床试验的“项目设计”、“医院选择”和“试验协调”,成了躲不过的拦路虎。
目前中国医疗器械上市的临床评价路径主要有三种:“免临床”、“同品种比对”和“临床试验”。第一种,“免临床”路径,国家严格规定了《免于进行临床试验的医疗器械目录》,大多为成熟度高、风险较低的常见器械,创新器械基本无缘;第二种,“同品种比对”路径,政策要求必须与已上市的同品牌前代产品或其他品牌类似产品,进行经验数据和产品资料的严格比对。但创新医疗器械的新颖性,决定了其一般不会有同品牌前代产品;而激烈的市场竞争,也决定了即使有其他品牌类似产品上市,这些品牌也不会授权别家企业使用自己的经验数据和产品资料(NMPA要求同品种比对必须获得被比对产品厂家的授权)。
如此一来,创新器械的临床评价,就只剩下了华山一条路,也即上文提到的了第三种路径:临床试验。经验表明,在创新医疗器械临床试验的时间表上,“项目设计”、“医院选择”和“试验协调”,每个阶段都存在很大不确定性。
临床试验“项目设计”之难,基于产品是否“够新”,也有“简单模式”和“困难模式”之别。“简单模式”,主要对应"Me-better"类微创新产品。在"Me-better"产品上市之前,往往已有其他品牌的类似产品上市。尽管由于前述原因,这类“Me-better”产品很难走“同品种比对”临床路径,但在走“临床试验”路径之时,仍可以对照已上市产品,比较双方临床试验结果。如此一来,临床试验的项目设计就会简单很多,很可能2-3家医院,几十例病例即可,一般只要大半年就能完成。与之相对,“困难模式”主要对应“Me-only”类创新产品,市面上没有先例产品可以作为参照。此种情形下,临床试验项目需要更加完善的设计,所需医院数和病例数均大幅增加,在近十家医院,进行百例以上的试验,前后持续2-3年,几乎成了常规操作。
“医院选择”之难,在于临床试验资质的稀缺,以及伦理审查流程的冗长。一方面,全国有医疗器械临床试验机构资格的医院,只有固定的几十家,平均每省只有1-2家,僧多粥少,医院相对企业处于绝对强势地位。另一方面,在医院开展临床试验需要“过伦理”,而每家医院,都有固定的伦理委员会审议流程。开会周期,多者每年五六次,少者每年一两次,且每次开会能审议的项目数量十分有限,这次排不上,只能等下次。如果试验碰巧是前述的“困难模式”,涉及到近十家医院的伦理审批,每家审批周期都要4-5个月,再加上每家医院伦理委员会开会时间不同,前后交错,光“过伦理”这一项工作,战线就可能迁延数月乃至一年以上,对企业的时间、人力和医院关系有都很大挑战。
“试验协调”之难,在于如何平衡各个相关方的利益,“让所有人满意”。临床试验的推进是个系统工程,涉及的利益相关方很多。除了前述的伦理委员会,还有患者、临床医生、科室主任、医工处、设备科、院管理层等,每一个环节运转不畅,都可能阻碍试验项目如期推进。例如,即使试验项目获得了伦理委员会的批准,还得靠设备科协调设备入院;即使设备成功入院,如果临床医生将之束之高阁,试验入组照样缓慢;即使临床医生也有动力尝试,还需要患者签署知情同意书,同意在治疗种使用试验器械。复杂的利益关系,必然会导至项目出现剪不断、理还乱的推诿、扯皮、潜规则。如果企业无法做到有效协调,项目进度拖延十天半月,甚至半年一年,都是常有之事。
陷阱二:物价审核困难
产品注册虽然冗长,但国家近年来针对创新医疗器械,力推注册流程的简化,曙光依稀可见。但是在创新医疗器械获得注册证后,“做物价”,也即“医疗服务收费审核”,又成了创新器械爆发式增长的最大阻碍。
所谓物价,也即“医疗服务项目收费标准”,是医院就某项医疗服务,可以向支付方收取的打包价格。该价格由“医院成本”和“医院利润”构成,其中“医院成本”一般包括以下几项: 一次性耗材成本、劳务成本、水电气及折旧成本、维修成本等。其中,如果该医疗服务项目需要使用某种特殊的高值耗材,不便添加到整体打包收费之中,还可以设定“除外耗材单独收费”,也即在收取医疗服务项目费用的同时,加收一笔“除外耗材费用”。
“做物价”,为什么重要?因为它直接决定了医院和科室,对某种创新医疗器械的接纳程度。众所周知,中国的公立医院自负盈亏,科室独立核算。对于某种创新医疗器械,只有其所对应的医疗服务项目获得了“物价审批”,医院和科室才能合法地向支付方收费,开展相应服务、使用对应器械,获得正向现金流,增加营收,带来利润;反之,如果该器械和对应的医疗服务项目没有获得"物价审批",医院和科室不能收费,只能“自己掏钱”开展相应服务、使用对应器械,导至出现负向现金流,增加成本,影响盈利。因此,只有在“有物价”的情况下,医院和科室才会有足够的经济动力,实现该创新器械的“上量”。
然而,做创新医疗器械的物价,谈何容易?行业内曾经有过测算,将一个创新医疗器械的物价做满全国,需要10年以上的时间。
比如某血浆类再生医学技术,在中国首个吃螃蟹的本土企业,是一家位于华东的大型医疗器械集团,其品牌认可度、区域覆盖及医院资源在中国均属顶级。即使坐拥这样的资源,自其产品2013年上市,截至2018年底,六年时间,也只在约10个省成功获批物价。
坐拥行业顶级资源的集团公司尚且如此,对于完全白手起家的单体创业企业,形势则更为严峻。比如国内某妇科射频消融技术企业,其产品属于全球领先、国内原创。用了约十年时间,才在约5个省获批物价。其个中艰苦,可想而知。
“做物价”之所以艰难,在于物价审批决策流程的去中心化。更具体而言,在于决策点分散、决策态度观望、决策相关方众多。
"决策点分散",根源在于国家级监管机构,仅对医疗器械的物价审批提框架性指导意见("全国医疗服务价格项目规范", 分为2001和2012两个版本,目前不同地区执行不同版本),而将具体审批权限和审批流程下放至各省。而各省又根据本省情况,或将审批权保留在省一级,或将审批权下放至地市级。由于各地政策不一,执行力度各异,医疗器械厂家需要针对不同省、市做工作,逐个公关,其工作量之大,工作内容之复杂,不言而喻。
"决策态度观望",则是决策流程去中心化的另一个必然结果。2012年,国家更新了“全国医疗服务价格项目规范”,但大多都省市都停留在了消化、吸收、观望新政策阶段,并未大幅开展“新物价”的审批。2015年底,发改委为了改变医疗服务项目“新增无门”的情况,专门发文要求各地加快受理新增医疗服务价格项目。其后,各地虽然逐步开始加快受理新的“物价申请”,但由于缺乏统一的审批流程,又恰逢医疗卫生相关部委架构和权责的重组,“新物价”的审批进度仍然十分缓慢。进入2018年底后,机构改革逐渐尘埃落定,新增物价才逐渐出现“松绑提速”的迹象。尽管如此,由于过去几年,各地积压了很多“新物价申请”,而每个省市每年获批的“新物价”又名额有限(比如北方某省,每年审批2次,一共只会批准20项左右),名额“僧多粥少”,获批仍然艰难,极度考验企业的政府关系管理能力。
"决策相关方众多",是做决策流程去中心化的另一体现。新物价的申请,首先要求医院设备科和临床科室已经采购了对应器械,然后从临床科室提出物价申请,到医院物价处、医疗器械管理委员会等部门预审,进而依各地流程,提交到发改委、医保局、物价局、卫健委中的一个部门或数个部门进行审批。流程复杂冗长,需要做工作的关联方众多,“跑断腿、说破嘴”成为常态。
历史经验表明,器械卖进医院、通过临床科室提出物价申请,一般需要数月甚至一年的酝酿协调期;医院相关部门一般会以每年2-4次的频率开会,预审相关物价申请,并决定将哪些申请提交政府审批;省内的物价申请,一般需要2-3家三甲医院同时提出,这也意味企业至少需要三线作战,三家医院同时做工作;对于不同省市,在不同历史时期,政府审批物价的频率也不一样。2018年以前,在某些省份,如浙江、北京、上海、广东等,2-3年才会审议一次物价。2018年底开始,各省才逐步提高审议频率,有的省份已经能够做到每年审议2次。如此算下来,将某省市的物价办妥,即使每个窗口期都踩到点上,至少也需要1.5-2.0年的时间。如果将“做物价”的目标扩展至全国大部分重点省份,再不小心错过几个关键节点,没有几年以上的时间打底,是不可能完成的。
陷阱三:临床接纳迟缓
临床应用,是医疗器械创新商业变现的终极途径。而前述的所有努力,无论是产品注册还是物价申请,都是为了临床应用做铺垫。而“临床接纳迟缓”,则是创新医疗器械在临床应用阶段,最严峻的挑战。
医疗器械的临床应用,可进一步细分为“入院”和“上量”两个阶段。创新医疗器械面临的“临床接纳迟缓”问题,在“入院”阶段,体现在“医院准入复杂”;在“上量”阶段,表现为“医生接受困难”。
医院准入之所以复杂,在于政府审批(大型医疗器械)和入院流程(对于所有医疗器械)两个方面。前者考验政府关系,后者需要渠道资源。
一方面,国家对于医疗设备的配置进行分级管理,甲类医疗设备(价格一般在3000万人民币以上)的配置规划,需要由国务院卫健委统一进行。乙类医疗设备(价格一般在1000-3000万人民币)的配置规划由省级卫健委进行,并报国务院卫健委核准实施。最典型的例子,莫过于达芬奇手术机器人。达芬奇机器人一经推出,便在中国的医生群体中获得了极高的人气和兴趣。但是由于属于大型医疗设备(2018年以前为甲类,2018年后从甲类调整为乙类),配置规划收到国家严格管控,这种超高的人气并没有能够有效地转化为装机量,使得“武器不足”的中国医生们只能在单机使用率上拼命提高,以弥补装机量的不足。截至2018年初,达芬奇只在中国装机了70余台,仅占其全球装机量(约4000台)的1.75%;但中国单台机器周转率是348台手术/年,是全球周转率的2倍。这些数据,一方面体现了中国医生不辞辛劳、尽可能满足患者手术需求的现实,一方面也折射出来装机量受制于政府审批的无奈。
另一方面,即使剩余的医疗器械,不需要政府的前置审批,若要顺利走完入院流程,也是对企业渠道资源的巨大考验。创新医疗器械一般不需集中招标采购,但复杂的入院和招标流程一样逃不掉。首先,企业需要针对相应临床科室进行需求挖掘和产品推广,启发科室需求(一般至少需要0.5-1.0年);进而,企业需要协助临床科室起草需求报告,提出器械采购申请;紧接着,企业需要与经销商合作,确保在相关申请顺利在医院管理层会议或是医疗器械委员会审议通过(一般一年2-4次);最后,企业和经销商需要协助设备科执行招标、交付产品(一般需要1-2个月,甚至更长)。粗算下来,完成单体大型三甲医院的入院,企业至少需要1.0-1.5年的时间预算,以及10万元左右的费用预算。如果把入院目标拓展至多家医院,考虑到不同医院在流程和时间线上的差异,企业的所需时间、费用和渠道资源势必成倍增加。
医生接受之所以困难,主要来源于三个方向的惯性,即品牌惯性、使用惯性和激励惯性。
医生的“品牌惯性”,尤其体现在对国产创新医疗器械的接受度上。具体而言,表现为两种错误观点:第一种错误观点,就是“国产 = 无创新”。尽管经过一二十年的发展,国产医疗器械取得了长足的进步,但观念的改变,总是落后于现实的变化,用户们的刻板印象仍然根深蒂固。诸如 "国产就是进口替代,"国产器械等于Me-too,最多是Me-Better"等观念仍然屡见不鲜,占据主流,短时间内难以扭转;第二种错误观点,就是“国产 = 纪律风险”。如果用了进口医疗器械,手术出了问题,就是产品问题;但如果用了国产医疗器械,手术出了问题,就是腐败问题。这样的刻板印象,导至了很多优秀国产创新医疗器械“墙里开花墙外香”的怪相。也即在国外市场广受欢迎,但却始终难以打入国内医院。
医生的“使用惯性”,顾名思义,就是医生已经习惯了现有产品的操作方法,如果需要从头学习某一种创新医疗器械的操作,医生有限的“精力成本”将成为不小的阻碍。这部分内容,笔者在之前的文章“洞见!搞医疗器械,就是要当这三种寡头”有详细阐述,在此不再赘言。
而医生的“激励惯性”,是造成“接受困难”的众多因素中,比较容易被忽略的一个。创新医疗器械之所以“新”,就是要替代现有产品。而现有产品通行多年,一般已经建立了非常有效的医生激励机制。其中,既包括非物质激励,也包括物质激励。对于非物质激励,例如学术会议、继续教育、研究支持等,行业已经形成了固定生态,而创新器械企业大多为创业公司,普遍缺乏相应资源;对于物质激励,现有产品往往已经建立了清晰的商业模式,将医院、科室和用户的利益串联其中,各有所得。而创新医疗器械的产品特点和作用机制,往往会打破既有商业模式,却又无法建立起成熟的新模式,导至在物质激励角度,无法有效提升创新产品的临床应用。
前文提到的血浆类再生医学产品在骨科领域的应用,就是一个典型例子。该产品最早在欧美市场出现,应用在心外科领域,后逐渐被扩展到骨科领域。在某些特定的亚适应症上,甚至出现了能够替代传统手术的可能性。但再生医学公司往往规模不大,与传统骨科植入物巨头相比,无法在上市后研究、技术培训和继续教育上匹配同样量级的资源,在“非物质激励”上落了下风;同时,由于现有植入物产品已经在骨科适应症的治疗上建立了清晰的商业模式,这些公司在“物质激励”上也占不到丝毫优势。以上两股合力,导至这类产品在骨科的适应症拓展在经历了短暂的“蜜月期”后,逐渐陷入增长瓶颈,难以进一步突破。
面对“产品注册”、“物价审核”和“临床接纳”三大商业化陷阱,创新医疗器械企业究竟应该如何应对,才能逃离“创新者的窘境”?
三、运用“底线思维”跨越创新陷阱
提出任何解决方案,我们都需要厘清问题的根源。创新医疗器械商业化之所以有“陷阱”,在于“产品注册”、“物价审核”和“临床接纳”三个步骤存在高度不确定性。而这种不确定性的根源,是创新医疗器械企业天然的颠覆性、新颖性,与监管机构、医疗机构内在的严谨性、滞后性之间的矛盾,而这种矛盾根植在各利益相关方的内在属性之中,几乎是不可调和的。
既然上述不确定性从客观上不可调和,无法消除,那么企业就必须从主观角度出发,“变不确定为确定”。一言以蔽之,就是“假装坏事必然会发生”。
这种“假装”有一个高大上的学名,就是“底线思维”。具体而言,就是设想最差情况可能是什么,假设该情况一定会发生,并坦然接受。凡事向“最差情况”准备,但努力争取最好的结果。
在中国商业领域,将底线思维应用到极致的,莫过于华为。2019年5月,经美国总统特朗普授意,美国商务部工业和安全局(BIS) ,将华为列入可能会威胁美国国家安全的实体名单(Entity List),禁止华为从美国购买技术或配件。然而,不过短短48小时,一封华为海思总裁宣布海思产品全部“备胎转正”的公开信,燃爆网络。原来,早在十多年前,“还是云淡风轻的季节”,华为就运用底线思维,做出了“极限生存的假设”,猜想未来某天,“所有美国的先进芯片和技术将不可获得”。正是基于这一底线思维,华为成立了华为海思半导体,为公司生存打造“备胎产品”,从而在“命运的年轮转到这个极限而黑暗的时刻,超级大国做出最疯狂的决定”的时候,华为仍然能够优步从容,“挽狂澜于既倒,确保公司大部分产品的战略安全,大部分产品的连续供应”。
那么,“底线思维”这把利剑,创新医疗器械企业应该如何运用?
总结起来, 一共分为三步:
-
第一步,设想最差情况
-
第二步,制定应对预案
-
第三步,考虑补救策略
第一步:设想最差情况
如何“设想最差情况”?企业需要通过对外反向对标、对内预设X因素的方法,输入、整合相关信息,针对上文提到“三大陷阱”,勾勒出“坏事必然会发生”的假想情况,并对“究竟能有多坏”做出极限预期。
首先,创新医疗器械企业要对外进行反向对标。众所周知,对标分析是一种以同行业、同细分领域优秀企业为标杆,从各个角度分析学习他人成功经验,以改善自身不足的分析方法。而反向对标,顾名思义,就是要分析研究同类医疗器械创新企业,在商业化道路上的失败经验,进而评估“这件事会不会发生在我们身上”。太阳底下没有新鲜事,如果同类的A公司在某一省物价审核上卡了3年毫无进展,那么假设自己公司1年之内就能全部搞定,就是非常危险的判断。
反向对标之后,创新医疗器械企业还需要对内预设X因素,为自己的计划打出“富余量”。创新企业通过自身经验和反向对标,设想“最差情况”之时,依据的是“已知的未知”(Known unknowns),也即企业明确了解到的、可能会发生的风险。但总有一些意外情况,是企业在做计划之时,无论穷尽怎样的手段,搜集再多的信息,都没法预见的。这种超出当前认知能力的“未知的未知”(Unknown unknowns),就是所谓的X因素,需要在“最差情况”中考虑在内,打出专门的“富余量”。
那么X因素的“富余量”,究竟应该如何匡算呢?通常来说,复盘是比较行之有效的方法。比如,某公司过去有2个临床试验,预计中的“最差情况”在现实中出现了。执行项目前,公司设想的“最差情况”,分别是24个月和30个月完成试验。但实践中的“最差情况”,临床试验分别耗时30个月和32个月才宣告完成。也就是说,这2个临床实验的X因素,分别导至了6个月和2个月的延期。那么,在设想第3个产品临床试验的“最差情况”时,公司就需要针对X因素,打出 4个月([ 2+6 ] ÷ 2 )的富余量。随着复盘次数的增加,公司认知能力中“未知的未知”越来越少,X因素对计划的影响也会越来越小,乃至最终忽略不计。
第二步:制定应对预案
明确“最差情况”后,公司应该进一步针对最差情况,“制定应对预案”:一旦最差情况真实发生,应该启动哪些事先制定的方案,把企业所受影响控制在最低限度?
经验表明,“最差情况”的“应对预案”,主要有风险应对和风险转嫁两大手段。
风险应对是最传统、最直接的手段。企业应该列出“最差情况”中设想到的相关问题,并一一列出每个问题对应的解决方法。解决方法的制定,既要考虑内部过往经验的总结,也要进行广泛的对标分析,引入其他企业的领先经验。
风险转嫁是目前比较受到忽视的一种手段。创新医疗器械大多来自于创业公司,风险承受能力小,稍有不慎,即有可能满盘皆输。如果能将“最差情况”中面临的风险,通过合理的机制,由其他机构共同承担,将极大提高企业的风险应对能力。
比较典型的风险转嫁方式有“引入合作”和“购买保险”两种。“引入合作”方面,企业可以考虑通过“与商业化巨头战略合作”、“利用第三方营销组织进行商业化”、“与有商业化转型需求的传统流通企业合作”等方式,借力打力,利用合作伙伴的能力和资源,跨越商业化陷阱。这一点笔者在文章“洞见|搞医疗器械,就是要当这三种寡头”中也有提及,此处不再赘言。
“购买保险”方面,“研发费用损失险”作为一种新兴的风险转嫁形式,正在获得各地政府不同程度的推动。2019年5月,中国人民保险(PICC)南京分公司与南京方生和医药科技有限公司签署研发费用损失险协议,约定如果方生和的差异化仿制药项目在保险期间未能按照立项预算完成,PICC将负责赔偿被保险人损失的研发费用(包括各种实验、测试费用等)。同样的模式能否应用在医疗器械企业,值得企业和政府借鉴探讨。
第三步,考虑补救策略
“应对预案”并非万灵丹。总有一些风险,是企业穷尽任何资源和手段,都无法完全消除的;总有一些问题,是决策者经验再丰富,也无法尽数解决的。在这种情况下,企业同样需要提前制定补救策略,作为备选方案,以平衡风险,达到“失之东隅,收之桑榆”的效果。根据笔者总结,这些替代方案,包括但不限于重新定义客户、进军海外市场、丰富商业模式等。
重新定义客户,尤其适用于同时存在于临床和非临床两个潜在使用场景的医疗器械。比如康复类、检验类器械等。典型例子是外骨骼机器人。在临床场景,外骨骼机器人被定义为医疗器械,目标客户群是临床医护人员,产品的使用以治疗、康复为目的,而前文叙述到的三大商业化陷阱,将一个不落地碰到;但在非临床场景,外骨骼机器人被定义为消费电子,主要消费者是非病人群体,产品的使用以助行、助力为主要目的,上述三大商业化陷阱,可以有效规避。虽然消费电子也有其独特的商业化挑战,但至少难度和周期上,都会比医疗器械小很多。如果企业的运营能够充分展现灵活性,在医疗器械商业化路径之外,探索非医疗器械的商业化路径,实现医疗器械 /消费电子双品牌或双产品战略,将是此类企业“应对预案”部分失效后,最有力的补救策略。
进军海外市场,有可能为国内商业化受阻的创新医疗器械企业,提前带来海外的营业收入。这一选择尤其适合“Me-better”类微创新医疗器械。其主要原因在于,海外注册(尤其是欧盟CE和美国FDA)对“Me-better”产品,较NMPA更为友好、快速;并且,尽管“Me-better”类产品海外上市也会遇到“物价问题”,但困难程度要比国内小很多。有鉴于此,很多受困于国内商业化陷阱的创新企业,纷纷提前开拓海外市场,作为“Quick-win”的重要手段。比如,中国消化内镜耗材企业和泌尿外科耗材企业,其“Me-better”类微创新产品在研发完成后,往往会在海外市场率先完成产品注册和上市销售。其中某家典型企业,其所有产品,只有不到一半拿到了国内的注册证,但几乎全部拿到了海外注册证。而其海外市场的销售收入,占到了企业总收入的70%以上。
丰富商业模式,是指在传统的分销或直销模式之外,把眼光放宽至其他可能的商业模式,从而规避前述的商业化陷阱。其中,典型的手段包括“技术转让(license-out)”和“贴牌代工(Private labelling)”。由于行业特性和发展历史的原因,此类替代模式一般对创新药比较常见,创新医疗器械较少涉及,但最近已越来越受到行业的重视。例如,数问生物于2019年3月宣布将其qixiaNIPT产品授权给IVD国际巨头PerkinElmer(珀金埃尔默),在中国以外的市场进行商业化,从而打响了中国创新医疗器械企业,面向海外市场“License-out”的第一枪。
结语
管理实践表明,在决策过程中,“不确定的损失”相比“确定的损失”,反而会让企业决策者产生更多的决策焦虑,消耗更多的精力和资源。这种决策焦虑,也正是人们对创新医疗器械的商业化视作畏途,不敢轻易触碰的重要原因之一。
然而,如果决策者能够运用底线思维,做一个“反向阿Q”,把“不确定的损失”全部转变为“确定的损失”,那么在创新医疗器械的商业化之路上,企业将几乎立于“不败之地”。毕竟,如果连“最差情况”,都在计划之内,连“最差情况”带来的商业回报都在可接受范围,那么企业从容跨过创新医疗器械商业化的“三大陷阱”,绝非天方夜谭。毕竟,“如果失望都惧我,那我还惧什么?”
来源:小桔灯网


